Medizin
Nr. 4 • April 2014
11
Sichelzellkrankheiten
Epidemiologie
In Deutschland leben 2013 schät-
zungsweise 1000–1500 Kinder und
Erwachsene mit Sichelzellkrankheit,
überwiegend in industriellen Bal-
lungsgebieten und Städten wie Berlin,
Hamburg, Stuttgart, Düsseldorf, Mün-
chen, Frankfurt. Die mediane Lebens-
erwartung von Patienten mit homo-
zygoter Sichelzellerkrankung (HbSS)
beträgt zwar 40–50 Jahre, ihr Leben
ist aber durch eine erhebliche Morbi-
dität gekennzeichnet [11].
Pathophysiologie
Das infolge einer Mutation im β-Glo-
bin-Locus auf Chromosom 11 verän-
derte Hämoglobin S (HbS) beeinflusst
Form, Oberflächeneigenschaften und
Lebensdauer der Erythrozyten und ist
somit die Basis der drei Phänomene,
welche die Erkrankung kennzeichnen:
Vaso-Okklusionen in allen Organen,
hohes Infektions-Risiko durch die
funktionelle Asplenie (Folge der Vaso-
Okklusionen) und chronische hämo-
lytische Anämie. Vaso-Okklusionen
entstehen durch ein komplexes
Zusammenspiel von veränderten
Erythrozyten, Endothelverletzungen,
Plasma-Proteinen, Thrombozyten
und Granulozyten. Im Knochenmark
ablaufende Vaso-Okklusionen mani-
festieren sich als Schmerzkrisen.
Diagnose
Zur optimalen Betreuung der Patien-
ten ist es unerlässlich, eine präzise
Diagnose zu stellen. Die Diagnose
wird durch eine Hb-Analyse gestellt.
Neben der am häufigsten vorkom-
menden homozygoten Erkrankung
HbSS gibt es die compound-hetero-
zygoten Formen HbS-β-Thalassämie,
HbSC, HbSD, HbS Lepore, HbSOArab,
die sich erheblich in den klinischen
Ausprägungen unterscheiden. Diese
Formen der Erkrankung entstehen
dadurch, dass von einem Elternteil
die HbS-Anlage, vom anderen eine
der anderen relevanten β-Globin-
Mutationen, wie z.B. HbC, vererbt
wird. Sehr häufig, vor allem in Zen-
tralafrika, ist die Koexistenz einer
HbSS-Homozygotie mit einer α-Tha-
lassämie. Heterozygote HbS-Träger
sind asymptomatisch mit einer Aus-
nahme: ca. 4% der HbS-Träger
entwickelt eine schmerzlose Makro-
hämaturie durch Papilleninfarkte. Die
HbS-Heterozygotie verändert das
Blutbild nicht.
Klinik
Zur Behandlung von Patienten mit
Sichelzellkrankheit wurden vom
Sichelzell-Informationszentrum für
Deutschland ein Leitfaden und von
der Deutschen Gesellschaft für Häma-
tologie und medizinische Onkologie
(DGHO) eine Leitlinie herausgegeben
[8, 9].
Akute Komplikationen
Die häufigste Krankheitsmanifesta-
tion sind
Schmerzkrisen
, die bei
Erwachsenen in Becken, Sternum,
Wirbelkörpern, Rippen und gelenk-
nahen Anteilen der langen Röhren-
knochen lokalisiert sind. Sichelzellpa-
tienten müssen sowohl Schmerzmit-
tel der Stufe I (Novaminsulfat, Ibupro-
fen) als auch der Stufe II (Tramadol)
zur Hand haben, um Schmerzkrisen
primär zu Hause beherrschen zu kön-
nen. Außerdem müssen sie über die
wichtigsten Faktoren, die Schmerz-
krisen auslösen können, informiert
werden: Schwimmen in kaltem Was-
ser, Dehydrierung, Alkohol, Rauchen.
Häufige Schmerzkrisen sind eine
Indikation zur Behandlung mit
Hydroxycarbamid.
Die zweithäufigste Ursache stationär-
er Aufnahmen ist das
akute Thorax-
Syndrom (ATS)
, das durch Fettem-
bolien aus dem Knochenmark oder
Infektionen entstehen kann, aber
auch durch Überwässerung und
Hypoventilation bei schlecht über-
wachter Schmerztherapie [17]. Klini-
sche Zeichen eines ATS sind: Thorax-
schmerzen, Fieber, Tachypnoe.
Sichelzellpatienten haben ein sehr
hohes
Sepsis-Risiko
durch Pneumo-
kokken und (v.a. im Erwachsenenal-
ter) durch gramnegative Keime.
Impfungen gegen Pneumokokken
(z.B. PCV-13, PSV-23) sowie jährlich
Influenza (das Influenza-Virus macht
die Mukosa durchlässig für Pneumo-
kokken) werden empfohlen [10].
Milzsequestrationen
(plötzliches
Versacken eines Teils des zirkulieren-
den Blutvolumens in der Milz)
ereignen sich bei Patienten mit HbSC
bzw. HbS-β-Thalassämie bis ins
Erwachsenenalter [12]. In den meis-
ten Fällen ist eine einmalige Transfu-
sion notwendig.
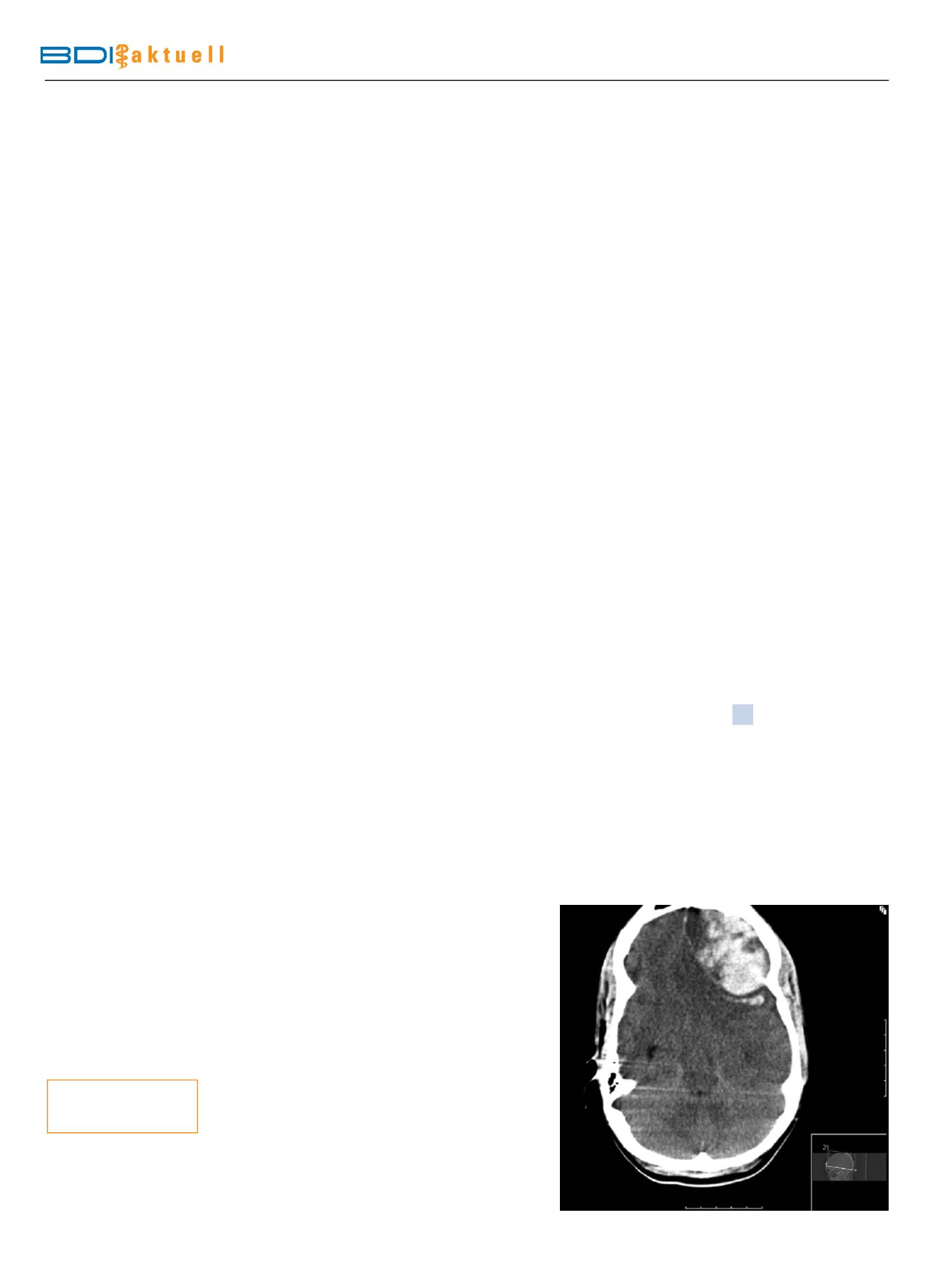
Akute ZNS-Ereig-
nisse
sind ab der 3. Dekade über-
wiegend ZNS-Blutungen (Abb. 1).
Ab dem 40. Lebensjahr werden ZNS-
Infarkte wieder häufiger. Nach einem
ZNS-Infarkt ist ein chronisches Trans-
fusionsprogramm indiziert um weit-
ere Infarkte zu verhindern.
Während einer Schwangerschaft kön-
nen sich bei Sichelzellpatientinnen
vermehrt Schmerzkrisen bzw. ATS
entwickeln, vor allem, wenn Hydroxy-
carbamid eingenommen wurde, das
in der Schwangerschaft abgesetzt
werden muss [7].
Chronische Probleme und Organschäden
Je älter Sichelzellpatienten werden,
desto häufiger haben sie, zusätzlich
zu den plötzlich auftretenden
Schmerzkrisen, chronische Schmer-
zen [2, 6]. Einige Ursachen können
gezielt angegangen werden: Deckplat-
teneinbrüche der Wirbelkörper (Phys-
iotherapie, Rückenschulung), aseptis-
che Nekrosen von Hüft- oder Hume-
rusköpfen oder gelenknahe Nekrosen
im Kniebereich (Anbohren der nekro-
tischen Areale). Bei Ausschluss dieser
Ursachen ist eine Dauermedikation
mit niedrigdosierten Retard-Opiaten
indiziert [1]. Bei chronischen Schmer-
zen ungewöhnlicher Lokalisation
(kleine Gelenke) muss an das Vor-
liegen einer zweiten Erkrankung wie
z.B. einer rheumatoiden Arthritis
gedacht werden [21].
Das häufigste chronische renale Prob-
lem ist neben der bei allen Sichelzell-
patienten vorhandenen Hyposthen-
urie die chronische glomeruläre
Nephritis, die sich durch eine Protein-
urie ankündigt. Sichelzellpatienten
haben wegen der hohen glomerulären
Filtrationsrate ungewöhnlich niedrige
Kreatinin-Werte und ein Kreatinin
von 0,8 mg/dl bedeutet bei ihnen
bereits den Beginn einer chronischen
Niereninsuffizienz.
Etwa 6–9% der älteren (> 40 Jahre)
Sichelzellpatienten haben einen pul-
monalen Hochdruck [16].
Viele Sichelzellpatienten, vor allem,
wenn sie über ethnische Grenzen hin-
weg transfundiert werden, entwick-
eln Antikörper gegen seltene Blut-
Untergruppen-Antigene, die weitere
Transfusionen problematisch bis
unmöglich machen, da keine passen-
den Konserven gefunden werden.
Besondere Beachtung verdienen
Patienten mit der Sichelzellkrankheit
HbSC. Bei ihnen muss ab dem 10.
Lebensjahr jährlich die Retina unter-
sucht werden, um eine proliferative
Retinopathie früh zu entdecken, mit
Laser-Koagulation zu behandeln und
die Patienten vor dem Erblinden zu
bewahren. Die bei Sichelzellpatienten
ungewöhnlich hohen Hb-Werte
(> 10 g/dl) stellen ein hohes Risiko für
Schwindel-Attacken, Hörstürze und
gehäufte Schmerzkrisen dar. In einer
solchen Situation bzw. vor Langzeit-
flügen (> 6 Stunden) sind Aderlässe
bei Patienten mit einem Hb > 11 g/dl
indiziert [15]. Bei HbS-β-Thalassämie-
Patienten kann sich bei Jugendlichen
oder Erwachsenen ein Hypersplenis-
mus mit Panzytopenie entwickeln.
Die Therapie der Wahl ist die
Splenektomie.
kurzgefasst
Sichelzellpatienten müssen
immer ausreichend Analgetika zu
Verfügung haben. Bei Thoraxschmer-
zen müssen sie stationär aufgenom-
men werden, um ein beginnendes
ATS nicht zu übersehen. Jedes Fieber
ungeklärter Ursache muss an eine
Sepsis denken lassen und entspre-
chend behandelt werden. Bei chroni-
schen Schmerzen sind aseptische
Nekrosen von Femur, Humerus und
Knien auszuschließen. Mindestens
jährliche Erhebung des Urinstatus und
Die Hämoglobinkrankheiten (Sichelzellkrankheiten und Thalassämien) sind weltweit die häufigsten erb-
lichen Erkrankungen. In Deutschland liegt die Häufigkeit der Trägerschaft in der Gesamtbevölkerung bei
4,5%, d.h. es gibt ca. 400 000 asymptomatische Träger dieser Erkrankungen [12]. Etwa 5 Millionen der
15 Millionen Menschen mit Migrationshintergrund in Deutschland kommen aus Ländern, in denen
Sichelzellkrankheiten und Thalassämien häufig sind (Tab. 1): Zentralafrika, Süd-Ost-Türkei, Mittlerer
Osten, Süd-Italien, Griechenland [18]. In Deutschland wissen die meisten Träger nicht um ihre Träger-
schaft, da es hier kein systematisches Screening gibt. Auch nicht bei der Erstuntersuchung einer Schwan-
geren aus einem Risikoland. Es sollte Routine werden, bei Schwangeren aus Risikoländern auf Thalassä-
mie oder Sichelzellkrankheit zu untersuchen und eine pränatale Diagnostik anzubieten, sollte der Partner
ebenfalls Träger sein [22]. In den Nachbarländern Deutschlands ist dieses Screening längst Routine.
Übersicht
Anämien und Hämoglobinkrankheiten
bei Patienten mit Migrationshintergrund
3 Blum RH, Carter SK, Agre K. A clinical
review of bleomycin – a new antineo-
plastic agent. Cancer 1973; 31: 903-914
4 Chen YB, Rahemtullah A, Breeden E et al.
Bleomycin-induced flagellate erythema.
J Clin Oncol 2007; 25: 898-900
5 Fernandez-Obregon AC, Hogan KP, Bibro
MK. Flagellate pigmentation from intra-
pleural bleomycin. A light microscopy
and electron microscopy study. J Am
Acad Dermatol 1985; 13: 464-468
6 Haas N, Vogt R, Sterry W. [Shiitake der-
matitis: flagellate dermatitis after eating
mushrooms]. Hautarzt 2001; 52: 132-
135
7 Hanada K, Hashimoto I. Flagellate
mushroom (Shiitake) dermatitis and
photosensitivity. Dermatology 1998;
197: 255-257
8 Jong SC, Birmingham JM. Medicinal and
therapeutic value of the shiitake
mushroom. Adv Appl Microbiol 1993;
39: 153-184
9 Kukla LJ, McGuire WP. Heat-induced
recall of bleomycin skin changes. Cancer
1982; 50: 2283-2284
10 Mahmoud BH, Eide MJ. Bendamustin-
induced "flagellate dermatitis". Derma-
tol Online J 2012; 18: 12
11 Maier T, Herzinger T. Linear dermatitis
due to shiitake mushrooms. Hautarzt
2007; 58: 1021-1022
12 Mak RK, Wakelin SH. Shiitake dermati-
tis: the first case reported from a Euro-
pean country. Br J Dermatol 2006; 154:
800-801
13 Moulin G, Fiere B, Beyvin A. [Cutaneous
pigmentation caused by bleomycin].
Bull Soc Fr Dermatol Syphiligr 1970; 77:
293-296
14 Nakamura T, Kobayashi A. [Toxicoder-
mia cause by the edible mushroom shii-
take (Lentinus edodes)]. Hautarzt 1985;
36: 591-593
15 Tallon B, Lamb S. Flagellate erythema
induced by docetaxel. Clin Exp Dermatol
2008; 33: 276-277
16 Wang X, Xu X, Zhang L. Thermally
induced conformation transition of tri-
ple-helical lentinan in NaCl aqueous
solution. J Phys Chem B 2008; 112:
10343-10351
17 Ziemer M, Goetze S, Juhasz K et al. Fla-
gellate dermatitis as a bleomycin-speci-
fic adverse effect of cytostatic therapy: a
clinical-histopathologic correlation. Am
J Clin Dermatol 2011; 12: 68-76
C. Schummer 1 , Y. Winkler 2 , J. Tittelbach 1 ,
M.-O. Grimm 2 , P. Elsner 1
1 Klinik für Hautkrankheiten, Universitäts-
klinikum Jena
2 Klinik für Urologie, Universitätsklinikum
Jena
Korrespondenz
Dr. med. C. Schummer
Klinik für Hautkrankheiten,
Universitätsklinikum Jena
Erfurter Straße 35
07743 Jena
Telefon: 03641-937365
Fax: 03641-937364
eMail:
-
jena.de
,
Der Beitrag ist erstmals erschienen in der
Deutschen Medizinischen Wochenschrift
(Dtsch Med Wochenschr 2014; 139: 84-
86). Alle Rechte vorbehalten.
Abb.1
Epi- und subdurale Blutung bei 19-jährigem Patienten mit Sichelzellerkrankung.


